
Key Concepts
In a dynamic or kinetic sense, chemical equilibrium is a condition in which a chemical reaction is occurring at equal rates in its forward and reverse directions, so that the concentrations of the reactant and product substances do not change with time. In a thermodynamic sense, it is the condition in which there is no tendency for the composition of the system to change. From the viewpoint of statistical mechanics, the equilibrium state places the system in a condition of maximum freedom (or minimum restraint) compatible with the energy, volume, and composition of the system. The statistical approach has been merged with thermodynamics into a field called statistical thermodynamics; this merger has been of immense value for its intellectual stimulus, as well as for its practical contributions to the study of equilibria. See also: Chemical thermodynamics; Statistical mechanics
Video
Chemical equilibrium of NO2 and N2O4. (Copyright © McGraw-Hill Education)
Because thermodynamics is concerned with relationships among observable properties, such as temperature, pressure, concentration, heat, and work, the relationships possess general validity, independent of theories of molecular behavior. Because of the simplicity of the concepts involved, this article utilizes the thermodynamic approach.
Chemical potential
Thermodynamics attributes to each chemical substance a property called the chemical potential, which may be thought of as the tendency of the substance to enter into chemical (or physical) change. Although the absolute chemical potential of a substance cannot be measured directly, differences in chemical potential are measurable. The units of chemical potential are joules per mole. See also: Potentials
The importance of the chemical potential lies in its relation to the affinity or driving force of a chemical reaction. It is useful to consider the general reaction (1),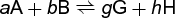 (1)
(1)
and let μA be the chemical potential of substance A, μB be the chemical potential of B, and so on. Then, according to one of the fundamental principles of thermodynamics (the second law), the reaction will be spontaneous (left to right) when the total chemical potential of the reactants is greater than that of the products. Thus, for spontaneous change (naturally occurring processes), notation (2)
applies. When equilibrium is reached, the total chemical potentials of products and reactants become equal; thus Eq. (3)
holds at equilibrium. The difference in chemical potentials in Eqs. (2) and (3) is called the driving force or affinity of the process or reaction; the affinity is zero when the chemical system is in chemical equilibrium. See also: Thermodynamic principles
For reactions at constant temperature and pressure (the usual restraints in a chemical laboratory), the difference in chemical potentials becomes equal to the Gibbs energy change ΔrG for the process in Eq. (4).
The decrease in Gibbs energy represents the maximum net work obtainable from the process. When no more work is obtainable, then the system is at equilibrium. Conversely, if the value of ΔrG for a process is positive, some useful work will have to be expended upon the process, or reaction, in order to make it proceed from left to right; the process cannot proceed naturally or spontaneously. The term “spontaneously” as used here implies only that a process can occur. It does not imply that the reaction will actually occur. Thus, the reaction between hydrogen and oxygen is a spontaneous process in the sense of the term as used here, even though a mixture of hydrogen and oxygen can remain unchanged for years unless ignited or exposed to a catalyst. See also: Free energy
Because, by definition, a catalyst does not undergo a net chemical change during the reaction, its chemical potential does not appear in Eqs. (2), (3), and (4). A catalyst, therefore, can contribute nothing to the driving force of a reaction, nor can it, in consequence, alter the position of the chemical equilibrium in a system. See also: Catalysis and catalysts
In addition to furnishing a criterion for the equilibrium state of a chemical system, the thermodynamic method goes much further. In many cases, it yields a relation between the change in chemical potentials (or change in Gibbs energy) and the equilibrium concentrations of the substances involved in the reaction. To do this, the chemical potential must be expressed as a function of concentration and possibly other properties of the substance.
The chemical potential μ is usually represented by Eq. (5),
where R is the gas constant, T is the absolute temperature, x is the concentration of the substance, γ is the activity coefficient of the substance, and μ0 is the chemical potential of the substance in its standard state. See also: Concentration scales
For substances obeying the laws of ideal solutions (or ideal gases), the last term, RT ln γ, is zero (γ = 1), because it is a measure of the deviation from ideal behavior caused by intermolecular or inter-ionic forces. An ideal solution would then be a solution for which RT ln γ is zero (γ = 1) over the whole concentration range. For real solutions, the state standard (where γ is unity) is generally chosen as the pure substance (mole fraction = 100 kilopascals and the temperature of interest) for solids and solvents, and as the hypothetical ideal gas or the unit concentration solute in solution at 100 kilopascals and the temperature of interest. Although the activity coefficient γ is dimensionless, its numerical values depend upon the particular concentration scale x with which it is associated.
Although the choice of concentration scales is a matter of convenience, the following are conventionally used: x = p = partial pressure, for gases; x = c (or m) = molar concentrations, respectively, for solutes in electrolytic solutions; x = mole fraction, for solids and solvents. When the choice is not established by convention, the mole fraction scale is preferred.
Activity and standard states
It is often convenient to utilize the product, γx, called the activity of the substance and defined by a = γx. The activity may be looked upon as an effective concentration of the substance, measured in the same units as the concentration x with which it is associated. The standard state of the substance is then defined as the state of unit activity (where a = x = 1) at 100 kilopascals and the temperature of interest and is characterized by the standard chemical potential μ0. Clearly, the terms μ0, γ, and x are not independent; the choice of the activity scale serves to fix the standard state. For example, for hydrochloric acid, the standard state for the solute (HCl) would be an (hypothetical) ideal 1 molar solution, and for the solvent (H2O) the standard state would be pure water (mole fraction = 1), both at 100 kilopascals and the temperature of interest. The reference state would be an infinitely dilute solution; here the activity coefficients would be unity for both solute and solvent. For the vapor of HCl above the solution, the standard state would be the hypothetical gaseous state at 100 kilopascals partial pressure. For gases, the term “fugacity” is often used instead of “activity.” See also: Activity (thermodynamics); Fugacity
Equilibrium constant
If the general reaction in Eq. (1) occurs at constant temperature T and pressure P when all of the substances involved are in their standard states of unit activity, Eq. (4) would become Eq. (6).
The quantity Δr G0 is known as the standard Gibbs energy change for the reaction at the temperature and pressure for the chosen standard states. (Standard state properties are commonly designated by a superscript zero, as in Δr G0, μ0.) Since each of the standard chemical potentials (μi0) is a unique property determined by the temperature, pressure, standard state, and chemical identity of the substance concerned, the standard free energy change ΔG0 is a constant characteristic of the particular reaction for the chosen temperature, pressure, and standard states.
If, for Eq. (1), at constant temperature and pressure, the chemical potential of each substance is expressed in terms of Eq. (5), then the Gibbs energy change for the reaction, from Eq. (4), becomes Eq. (7)
in terms of the activities and the standard Gibbs energy change, Eq. (6). Equation (7) is often written in the form of Eq. (8),
where Q is the ratio of the activities of products to the activities of reactants, each activity bearing as an exponent the corresponding coefficient in the balanced equation for the reaction [as in Eq. (7)]. The standard Gibbs energy change Δr G0 serves as a reference point from which the actual Gibbs energy change Δr G can be calculated in terms of the activities of the reacting substances.
When the system has come to chemical equilibrium at constant temperature and pressure, Δr G = 0, from Eq. (3). Equation (7) then leads to the very important relation shown in Eq. (9),
where the value of K is shown as Eq. (10),
and the activities are the equilibrium values. The quantity K is called the equilibrium constant for the reaction. The equilibrium constant is a characteristic property of the reaction system, because it is determined uniquely at a given temperature in terms of the standard Gibbs energy change. The term −Δr G0 represents the maximum net work that the reaction could make available when carried out at constant temperature and pressure with the substances in their standard states. It should be clear from Eq. (6) that the magnitude of Δr G0 is directly proportional to the amount of material represented in the reaction in Eq. (1). Likewise, Eqs. (7) through (10) denote this same proportionality through the exponents a, b, g, and h in the terms K and Q. Naturally, the value of Δr G0, as well as K and Q, will depend upon the particular concentration scales and standard states selected for the system. Therefore it is essential that sufficient information be stated about a system to fully describe the chosen standard states.
Equations (8) and (10) can be combined in the form of Eq. (11),
sometimes denoted as the Lewis equation after G. N. Lewis, who was the first to formulate this equation. When Q for a specified set of conditions is larger than K (so that Δr G is positive), the proposed reaction cannot occur left to right. On the other hand, when Q is less than K, then the proposed process or reaction can occur. The equilibrium constant thus serves as a measure of the position of chemical equilibrium for a system. For a proposed process for which Q is greater than K, the reaction system would have to move away from its equilibrium state (impossible of its own accord) to proceed from left to right, and for a proposed process for which Q is less than K, the process would bring the system closer to its equilibrium state (as in all naturally occurring processes).
Instead of activities, values of concentrations and activity coefficients at equilibrium may be used to express the form of the equilibrium constant K in Eq. (12).
This gives the equilibrium constant as a product of two terms. The first term, involving concentrations, is directly measurable if the system can be analyzed at equilibrium. On the other hand, the activity coefficient term, as seen in Eq. (13),
is frequently difficult to evaluate.
Intensive studies of activity coefficients made upon a wide variety of chemical systems led to a number of simplifying principles and some useful theoretical treatments of the subject. For gases, the activity coefficients differ only slightly from unity for pressures up to a few atmospheres and often can be evaluated from equation-of-state data. For mixtures of nonelectrolytes, the values also are not far from unity in many cases. For solutions of electrolytes, the activity coefficients vary greatly with concentration, and in many cases approach unity only below a useful or even attainable concentration. The theoretical treatments of P. Debye, E. Hückel, and others have systematized the patterns of electrolyte behavior, making possible a reasonable estimate of the activity coefficients of electrolytes in many cases. See also: Electrolyte; Gas
In general, the function Kγ , Eq. (13), approaches unity as the composition of the system approaches ideal behavior, so in practice most equilibrium constants K are evaluated through some suitable extrapolation procedure involving Eq. (12). See the discussion following Eq. (5).
For many approximate calculations or when data for Kγ are scarce, it is common to express the equilibrium constant as the concentration term only; that is, Eq. (14)
is applied. Unless Kγ is a rather insensitive function of concentration, the constants (Kapp denotes an apparent or approximate value of K) obtained in this manner are not constant as the composition is varied, and even though approximately constant, may vary considerably from the true value of K. Although the practice of assigning Kγ a value of unity will often give useful results and is frequently the only expedient available, the results must be used with caution.
The kinetic concept of chemical equilibrium introduced by C. M. Guldberg and P. Waage (1864) led to the formulation of the equilibrium constant in terms of concentrations. Although the concept is correct in terms of the dynamic picture of opposing reactions occurring at equal speeds, it was not successful in coping with the problems of activity coefficients and cannot lead to the useful relations, Eqs. (4) and (11), in terms of an energetic criterion for the position of equilibrium. Conversely, the thermodynamic approach yields no relationship between the driving force at the reaction and the rate of approach to equilibrium.
The influence of temperature upon the chemical potentials, and hence upon the equilibrium constant, is given by the Gibbs-Helmholtz equation (15).
The derivative on the left represents the slope of the curve obtained when values of ln K for a reaction, obtained at different temperatures, are plotted against temperature. The standard enthalpy of reaction Δr H0 for the temperature T at which the slope is measured, is the heat effect which could also be observed by carrying out the reaction involving the standard states in a calorimeter at the corresponding temperature and 100 kilopascals. See also: Calorimetry
For endothermic reactions, which absorb heat (Δr H0 positive), K increases with increasing temperature. For exothermic reactions, which evolve heat (Δr H0 negative), K decreases with increasing temperature and the yield of products is reduced. A more useful arrangement of Eq. (15) is shown in Eq. (16).
Plots of ln K against 1/T are nearly linear for reactions where the value of Δr H0 changes slowly with temperature. Hence, over small temperature ranges, Eq. (16) becomes, in integrated form, Eq. (17).
This relation is much used for calculating heats of reaction from two equilibrium measurements or for determining an equilibrium constant K2 at temperature T2 from values of K1, and Δr H0.
For accurate work, or for extending the calculations over a wide range of temperature, Δr H0 must be known as a function of temperature before Eq. (15) or (16) can be integrated. When sufficient heat capacity data are available, Kirchhoff's equation, involving the difference in heat capacities between the products and reactants, Eq. (18), may be combined with Eq. (15) to yield Eq. (19).
In these equations, the coefficients α, β, and δ are determined from heat capacity data; the constants Δr H00 and I require knowledge of one value of Δr H0 and one value of K, or values of K at two temperatures. See also: Heat capacity; Thermochemistry
Homogeneous equilibria
These equilibria involve single-phase systems: gaseous, liquid, or solid solutions.
A representative gas-phase equilibrium is the ammonia synthesis shown in Eq. (20),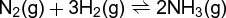 (20)
(20)
where (g) denotes a gas-phase species. The most natural concentration measures are mole fraction or partial pressure. The partial pressures pi are defined in terms of mole fractions xi , and the total pressure P by pi = xi P. For low pressures, where activity coefficients are practically unity, Eq. (21) holds. Equation (21) is a special case of Eq. (22),
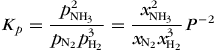 (21)
(21)where Δn is the change in the number of moles of gases [Δr = −2 for Eq. (20)]. The mole fraction equilibrium constant Kx is pressure dependent, but Kp is independent of pressure. Consequently, an increase in total pressure P must lead to an increase in Kx in this case. If the increase in total pressure is due to a decrease in volume of the system, then the result will be an increased yield of products (NH3). An increase in pressure brought about by injection of an inert gas into a constant volume system would not affect the partial pressures of the reacting gases nor the ultimate yield of products. The value of Kx , and thus that of ΔrGx , will be affected by a change in total pressure owing to a change in the net work of mixing and unmixing the gases. The reaction described by Eq. (20) is exothermic, so better yields will be obtained at lower temperatures. However, the rate of attainment of equilibrium in this case is very slow at low temperatures [Eq. (15)]. See also: Ammonia
A typical liquid-phase equilibrium is the deprotonation of acetic acid in water, reaction (23) and Eq. (24),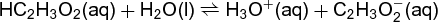 (23)
(23)
where (aq) denotes an aqueous-phase species and (l) denotes a liquid-phase solvent. In this particular case, the activity coefficient ratio Kγ is less than unity in dilute solutions. The activity of water aH2O ≃ xH2O at low molalities (≤1 m) is close to unity. For this reason, aH2O is commonly omitted in the formulation of the equilibrium constant expressions of this type, because aH2O ≃ 1. See also: Acetic acid; Ionic equilibrium
Heterogeneous equilibria
These are usually studied at constant pressure, because at least one of the phases will be a solid or a liquid. The imposed pressure may be that of an equilibrium gaseous phase, or it may be an externally controlled pressure.
In describing such systems, as for all reaction systems, the nature of each phase must be specified. In the examples to follow, s, l, g, aq, and soln identify solid, liquid, gaseous, aqueous, and nonaqueous phases, respectively. For solutions or mixtures, the composition is needed, in addition to the temperature and pressure, to complete the specification of the system; the nature of any solvent also must be specified.
In the equilibrium shown as (25),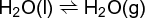 (25)
(25)
the relationship of Eq. (26)
holds. Here K ≈ p/x, the ratio of the vapor pressure p to the liquid mole fraction x. For pure water, the equilibrium constant is approximately equal to the vapor pressure of water at least at low pressures. When a small amount of solute is added, thereby decreasing the mole fraction of solvent, the vapor pressure p must be lowered to maintain equilibrium (Raoult's law). The effect of the total applied pressure P upon the vapor pressure p of the liquid is given by the Gibbs-Poynting equation (27).
Here Vl and Vg are the molar volumes of liquid and vapor, respectively. The equilibrium vapor pressure will increase as the total pressure is increased (both Vl and Vg are positive and the activity of the liquid increases with pressure). If the external pressure is applied to a solution by a semipermeable membrane, then an applied pressure can be found which will restore the vapor pressure (or activity) of the solvent to its standard state value.
For a solid, such as barium sulfate, BaSO4, which dissolves in water as shown in Eq. (28),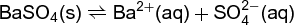 (28)
(28)
the equilibrium relationship is shown in Eq. (29).
When the solid state is pure, its activity is unity at 100 kilopascals. If the solid is extremely finely divided, then its activity is greater than unity; with this increase in the activity of the solid state, the solubility must increase to maintain equilibrium. On the other hand, inclusion of foreign ions in the crystal lattice (solid solution formation) lowers the activity of the solid state.
When a gas, such as CO2, is dissolved in a liquid, its equilibrium with the gas phase is as shown in Eq. (30).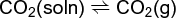 (30)
(30)
Equation (31),
Henry's law, represents the equilibrium constant for this equation, where m is the molality of dissolved CO2. When the gas dissociates in the liquid, as in reaction (32), Eq. (33)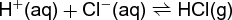 (32)
(32)
may be utilized. Here m is the molality of dissolved HCl.
Similarly, when a solute distributes itself between two immiscible phases, that is, nA(soln 1) An(soln 2), the equilibrium constant takes the form of Eq. (34).
An(soln 2), the equilibrium constant takes the form of Eq. (34).
The equilibrium concentrations ci in Eq. (34) reflect the relative solubilities of the solute in the two phases; since solubilities may vary widely, solvent extraction may provide an effective means for concentrating a widely dispersed solute. See also: Extraction
When components form immiscible phases at equilibrium, each condensed phase will be a saturated solution; complete immiscibility is impossible in principle, because the chemical potential of any component must be the same in all phases. The separation of a liquid system into two liquid phases is a manifestation of the nonideality of the solutions. For practical purposes, however, many solids may be regarded as immiscible because of the stringent requirements associated with formation of solid solutions.
In reactions involving condensed and immiscible phases, for example, reaction (35),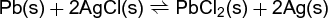 (35)
(35)
there can be no change in concentration of any phase during the reaction. In such a case the Q term in Eq. (11) will be constant, and ΔrG is fixed at a given pressure and temperature. Although a reaction is possible, there is at most one pressure at each temperature for which ΔrG could be zero. Such is the case in transition phenomena, or melting-freezing phenomena. From the phase-rule viewpoint, the system in reaction (35) has only one degree of freedom if reaction is possible, so once a value of P or T is chosen, the remaining variable (T or P) is fixed by nature at equilibrium. See also: Phase rule

