
The application of thermodynamic principles to systems involving physical and chemical transformations is carried out in order to (1) develop quantitative relationships among the identifiable forms of energy and their conjugate variables, (2) establish the criteria for spontaneous change, for equilibrium, and for thermodynamic stability, and (3) provide the macroscopic base for the statistical-mechanical bridge to atomic and molecular properties. The thermodynamic principles applied are the conservation of energy as embodied in the first law of thermodynamics, the principle of entropy production as embodied in the second law, and the principle of absolute entropy and its statistical thermodynamic formulation as embodied in the third law of thermodynamics.
Basic concepts
The basic goal of thermodynamics is to provide a description of a system of interest in order to investigate the nature and extent of changes in the state of that system as it undergoes spontaneous change toward equilibrium and interacts with its surroundings. This goal implicitly carries with it the concept that there are measurable properties of the system which can be used to describe adequately the state of the system, and that the system is enclosed by a boundary or wall which separates the system and its surroundings. Properties that define the state of the system can be classified as extensive and intensive. Extensive properties are proportional to the total mass of the system, whereas intensive properties are not. Typical extensive properties are the energy, volume, and numbers of moles of each component in the system, while typical intensive properties are temperature, pressure, density, and the mole fractions or concentrations of the components.
Extensive properties can be expressed as functions of other extensive properties, for instance, as in Eq. (1),
where the volume V of the system is expressed in terms of the internal energy U, the entropy S, and {ni}, the set of numbers of moles of the various components labeled by the index i. A suitable transformation procedure can be used to replace extensive variables by conjugate intensive variables. For example, the volume can be expressed as in Eq. (2 a) or (2 b).
Because temperature T and pressure P are particularly convenient variables to control and measure in chemical systems, the form of Eq. (2 b) is of great utility. All extensive thermodynamic properties X can be rewritten in this form, as Eq. (3).
Because all such properties are linear homogeneous functions of the mass, it can be shown that at a given temperature and pressure Eq. (4)
holds, where 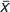 i is the partial molar value of the extensive property for the ith component. The value of i is given by Eq. (5),
i is the partial molar value of the extensive property for the ith component. The value of i is given by Eq. (5),
where the notation {ni}′ means that all amounts are constant, except the ith one involved in the derivative. The variable i is itself intensive.
Specification of boundaries
The concept of a boundary enclosing the system and separating it from the surroundings requires specification of the nature of the boundary and of any constraints that the boundary places upon the interaction of the system and its surroundings. Boundaries that restrain a system to a particular value of an extensive property are said to be restrictive with respect to that property. A boundary which restrains the system to a given volume V is a fixed, rigid wall. A boundary which is restrictive to one component of a system but not to the other components is called a semipermeable wall or membrane. A system whose boundaries are restrictive to energy and to mass of components is said to be an isolated system. A system whose boundaries are restrictive only to mass of components is called a closed system, whereas an open system has nonrestrictive walls and hence can exchange energy, volume, and mass with its surroundings. Boundaries can be restrictive with respect to specific forms of energy. Two important types are those restrictive to thermal energy but not work (adiabatic walls), and those restrictive to work but not thermal energy (diathermic walls).
First law of thermodynamics
Thermodynamics concerns the conversion of energy. The laws of thermodynamics differ from other scientific laws in that they are stated in terms of the impossibility of achieving certain types of energy transfers. The laws are usually stated in terms of the primary thermodynamic variables of temperature, energy, heat, work, and entropy.
Stated in terms of the primary thermodynamic functions of temperature, energy, and entropy, the laws of thermodynamics for any actual (that is, nonhypothetical) process are the first law: the total energy is conserved (that is, remains unchanged); the second law: the total entropy increases in any real process; and the third law: the absolute zero of temperature always cannot be reached in any real process.
Temperature
A device designed to measure temperature is called a thermometer. Familiar examples are the liquid-in-glass capillary, thermocouples, and resistance thermometers. All these devices have a property that varies monotonically with temperature. The fundamental thermometer is the constant-volume–ideal-gas thermometer, which is based on the fact that gases for which the constituent molecules do not combine or disassociate (that is, A + A 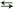 A2 or A2 2A) obey the ideal-gas equation (6)
A2 or A2 2A) obey the ideal-gas equation (6)
in the limit as P → 0, where P is the pressure of the gas, V is the volume, n is the number of moles of the gas, and R is the gas constant. With a constant-volume gas thermometer, the temperature of a fixed reference point, such as the melting point of lead metal, is measured using Eq. (7).
The pressure, PT, of the fixed mass of the gas at temperature T is measured for progressively smaller values of the mass of the gas. The resulting calculated values of temperature are extrapolated to zero pressure to obtain the thermodynamic temperature. The value of P273.16 K is the corresponding measured pressure of the gas when the thermometer bulb is in contact with water at its triple point. The triple point of water is the single most important reference point in thermometry. It is arbitrarily assigned a temperature of exactly 273.16 K simply in order to preserve a close numerical agreement with older temperature scales, such as the Celsius scale. See also: Triple point
The temperature scale defined by Eq. (7) is an absolute scale. Zero is the lowest possible temperature, and all measurable temperatures on this scale are positive. This temperature scale is called the absolute Kelvin temperature scale, and temperatures are expressed as kelvins. Celsius temperatures are related to Kelvin temperatures via Eq. (8).
The temperatures of fixed reference points obtained in the manner outlined above are, in turn, used to calibrate other, more convenient thermometers. See also: Absolute zero; Gas thermometry; Temperature; Thermometer
Energy, work, and heat
Energy is an abstract mathematical concept that is characterized by an energy function, U. Some other thermodynamic functions also are called energy functions (Gibbs energy, Helmholtz energy) and are chosen specifically as state functions that yield exact differentials in terms of experimentally convenient variables such as temperature and pressure. Energy is not a thing. There are no meters capable of the direct measurement of energy, yet there are instruments that measure electric current, gas flow, volume, mass, pressure, length, time, and temperature. These measurable properties are used to calculate energy changes. Determination of the amount of energy transferred from one system to another always involves measurements of the appropriate physical parameters, followed by calculations involving the appropriate energy formulas. See also: Energy
The principle of energy conservation amounts to the statement that, for any real process, the total energy of the system remains unchanged during the process. Energy can be transferred and transformed, but it cannot be created or destroyed. Work and heat are the two modes of energy transfer between systems. The transfer of energy as work requires the existence of an unbalanced force between a system and its surroundings. The transfer of energy as heat requires the existence of a temperature difference between the system and its surroundings. When a force F acts on a system and displaces the system by a differential amount dx, then the work done on the system is given by Eq. (9).
See also: Conservation of energy
The work w done on a system depends on the path along which the process is carried out, which must be specified before w can be calculated. When the path must be specified before the integral in Eq. (9) can be evaluated, then the integral is called a line integral. If the work done is independent of the path, then the force F is said to be conservative. A conservative force can always be expressed as the negative derivative of a potential energy, ϕ, with respect to the displacement, as in Eq. (10). Thus, for a conservative force, Eq. (11)
holds for the process. Equation (11) shows that, if the force is conservative, then the work done depends only on the end states (1 and 2) and is thus path-independent. See also: Work
If at any stage in the process energy is transferred as heat, then the forces acting on the system cannot be conservative, the value of w depends on the path, and the discipline that describes the process moves from classical mechanics to thermodynamics. For a general process involving both work and heat transfers, the first law of thermodynamics is given by Eq. (12).
See also: Heat
The energy function, U, is called the internal energy, and it is a thermodynamic state function. The differential of a thermodynamic state function is said to be exact because the value of ΔU = U2 − U1 for the process is path-independent, even if energy is transferred as heat and work in the process, as in Eq. (13).
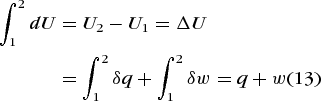 (13)
(13)See also: Differential equation; Internal energy
A key point is that for a given change in state (that is, 1 → 2) the value of U2 − U1 is independent of path, whereas the values of both q and w depend on the path. It is only the sum q + w that is independent of path. This statement constitutes the essence of the first law of thermodynamics. The following special cases are notable. If the process is adiabatic (that is, δq = 0), then δw is exact, because w = ΔU. If no work is done (that is, δw = 0), then δq is exact (pure heat transfer), because q = ΔU. See also: Adiabatic process
Reversible and irreversible processes
Changes in the state of the system can result from processes taking place within the system and from processes involving exchanges of mass or energy with the surroundings. After a process is carried out, if it is possible to restore both the system and the surroundings completely to their original states, then the process is said to be reversible; otherwise the process is irreversible. All naturally occurring spontaneous processes are more or less irreversible.
The internal energy of the system is given by the fundamental equation of state (14).
Processes which give rise to a change in U are then limited to those for which Eq. (15)
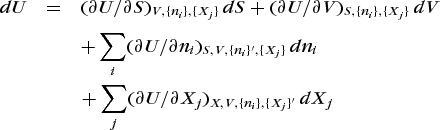 (15)
(15)holds. Because all the extensive state properties are linear homogeneous functions, the coefficients of the differential terms are themselves intensive functions and correspond in each case to the conjugate variable of the respective extensive variable. Their product is thus the differential work associated with the appropriate form of energy transfer. The table lists several forms of internal energy transfer, their conjugate pair of variables, and their corresponding work terms.
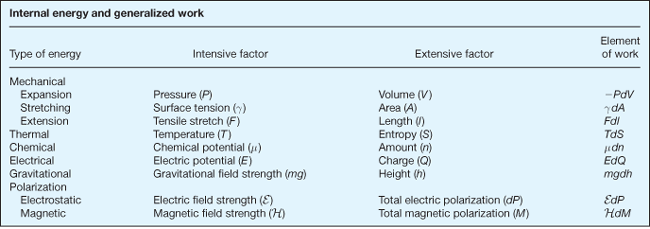
Equation (15) can be rewritten as Eq. (16),
where μi is the chemical potential of the ith component and Ij is the conjugate potential for Xj. The internal energy change given by Eq. (16) is dependent only upon the state properties of the system, and hence is independent of the process causing the change.
Enthalpy and heat capacity
A process involving only pressure-volume work may be described by Eq. (17).
Here, Pext denotes the externally applied pressure, the definition of pressure P = F/A = force/area is employed, and the volume element dV = (area of the base) × (vertical displacement) = A dx. Combination of Eqs. (12) and (17) yields Eq. (18).
The enthalpy H, a thermodynamic state function, is defined by Eq. (19), and differentiation of this equation yields Eq. (20).
Combination of Eqs. (18) and (20) yields Eq. (21), and, at constant pressure (dP = 0), Eq. (22)
holds. From this equation it follows that the addition or removal of energy as heat from a system at constant pressure is equal to the change in enthalpy of the system (ΔH = qp). See also: Enthalpy
The heat capacity of a system is a quantitative measure of the capacity of a system to take up or supply energy as heat. The greater the heat capacity of a system, the smaller the change in temperature produced by the input of energy as heat. The heat capacity is necessarily positive. The heat capacity Cx of a system at constant x (where x is a thermodynamic state function, such as pressure) is defined by Eq. (23),
where Cx(x, T) denotes that Cx is a function of both x and T. For a constant-pressure process, it follows from Eqs. (22) and (23) that Eq. (24)
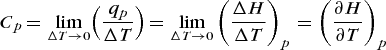 (24)
(24)holds. Also, from Eqs. (24) and (22), it follows that Eq. (25)
is valid. A large heat capacity indicates that the system can take up substantial amounts of energy as heat, owing to storage of the energy in the internal modes (translation, rotation, vibration, molecular dissociation) of the constituent particles. For example, the molar heat capacities of solid (s), liquid (l), and gaseous (g) water over the temperature range 250 to 500 K (−23 to 227°C or −10 to 440°F) are as follows:
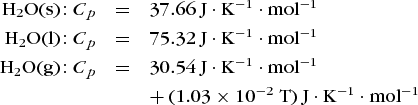
The roughly twofold greater heat capacity of liquid water relative to solid or gaseous water results from the fact that a substantial (roughly half) part of the heat input to liquid water is absorbed in breaking the hydrogen bonds between water molecules in liquid water (about 20 kJ/mol). Ice absorbs energy as heat below its melting point without breaking any hydrogen bonds and thus has a lower value of Cp than liquid water, whereas there are no hydrogen bonds in gaseous water and thus this mode of energy uptake is absent for the gas. See also: Heat capacity
Second law of thermodynamics
Most naturally occurring processes involve the transfer of energy as heat. The first law of thermodynamics places no restrictions on energy transfers other than that of conservation of energy. There are, however, other restrictions on energy transfer, namely:
1. Heat always flows spontaneously from higher- to lower-temperature systems.
2. Heat can be induced to flow from a lower- to a higher-temperature system only through the use of energy as work to drive the transfer.
3. When a system undergoes a change in thermodynamic state, then the system and its surroundings cannot both be restored exactly to their original states. All naturally occurring processes are in this sense irreversible.
A mathematical statement of the second law of thermodynamics is that the change in entropy of the system, dSsys, obeys inequality (26),
where T is the absolute thermodynamic temperature [measured in kelvins (K)]. The equality sign holds if the process is reversible, and the inequality sign holds if the process is irreversible. The entropy S is a thermodynamic state function, and it is expressed in units of joules per kelvin (J/K).
It is useful to consider a system and its surroundings between which energy can be transferred. If the system undergoes a change in state, then the total entropy change is given by Eq. (27).
If the process is adiabatic, then δqsys = δqsur = 0 and from Eq. (26) dSsur ≥ 0 and dSsys ≥ 0. Thus dStot ≥ 0, or ΔStot ≥ 0.
If the system undergoes an isothermal process, then T = Tsys = Tsur. From the second law, it follows that the system and the surroundings obey inequalities (28),
but qsys = −qsur and thus ΔStot ≥ 0. Note that in both cases there is a net entropy production if the process is irreversible, but the total entropy remains unchanged if the process is reversible.
All real processes involve some degree of irreversibility and thus lead to an increase in the total entropy. Entropy is not conserved, except in the hypothetical limiting case of a reversible process.
A thermodynamic system is said to be in an equilibrium state when it can no longer undergo any spontaneous (entropy-producing) processes. The increase in total entropy that occurs in an irreversible process is a consequence of the failure to fully exploit the potential of the system for performing work. When less than the maximum possible work is performed in a process, then net entropy production inevitably results. The total entropy can never decrease and, for a given energy and volume for a closed system, a maximum in entropy indicates the most stable configuration of the system, that is, equilibrium.
Because entropy is a state function, Eq. (29)
is satisfied. For a given change in state (1 → 2) the change in entropy of the system (but not the total entropy change, ΔStot = ΔSsys + ΔSsur) is independent of the path that the system takes from state 1 to state 2.
There are no entropy meters. Rather, entropy changes are calculated from measurable quantities, such as temperature, pressure, volume, and heat capacity. The total entropy of a system is a measure of the spatial and energy disorder (randomness) in a system.
Compared at the same pressure and temperature, liquids are more disordered than solids and gases are more disordered than liquids. This results follows from the fact that the molecules of a gas have more freedom to move around than the molecules of a liquid. The same holds true for the molecules or ions of a liquid as compared to those of a solid. The melting of a solid involves an entropy increase in the range 8–40 J · K−1 · mol−1. The temperature dependence of the entropy is found from Eqs. (26) and (24). From Eq. (26) it follows that qp = TΔS for an isothermal pure heat transfer (that is, δw = 0) at constant pressure. Substitution of this result into Eq. (24) yields Eq. (30). Thus, Eqs. (31) and (32)
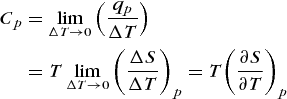 (30)
(30)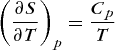 (31)
(31)are valid.
Because Cp is greater than 0, the entropy of a substance always increases with increasing temperature at constant pressure. The increase in the entropy of a substance that undergoes an increase in temperature at fixed pressure is a consequence of the increased thermal randomness of the molecules at the higher temperature. A greater amount of energy is more randomly distributed over the same number of particles. See also: Entropy
Gibbs energy and Maxwell relations
Combination of the first law, Eq. (12), with the second law, Eq. (26), yields Eq. (33).
If a process involves only pressure-volume work and occurs reversibly, then, applying Eq. (17), Eq. (33) becomes Eq. (34).
Because U and S are state functions, the values of ΔU and ΔS can be computed along a reversible path between the given initial and final states of the actual process, and the results for ΔU and ΔS for the system can be equated to those for the actual irreversible process. The values of q and w differ for the two paths, but not the values of ΔUsys or ΔSsys.
Equation (34) is the differential equation used to calculate ΔU. It expresses the change in the dependent variable in terms of changes in the independent variables S and V; that is, U = U(S, V). The variables S and V are not convenient independent variables because they are very difficult to control in the laboratory. More convenient independent variables for experimental work are pressure and temperature. Equation (34) can be transformed into a differential equation with P and T as independent variables through Eqs. (35).
For compactness, the thermodynamic energy state function G is defined by Eq. (36).
It can be used as a criterion for system equilibrium in terms of the more convenient variables, T and P. Here, H is defined by Eq. (19).
The function G is called the Gibbs energy after its creator, J. W. Gibbs; it plays a central role in chemical thermodynamics. From Eqs. (34) and (36), it obeys Eq. (37),
which gives the differential equation for dG, where G = G(T, P). Because dG is an exact differential, G is a state function, and from mathematics Eq. (38) holds. Comparison of Eqs. (38) and (37) leads to Eqs. (39) and (40).
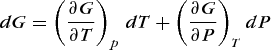 (38)
(38)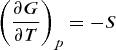 (39)
(39)Equation (39) shows that the temperature dependence of the Gibbs energy at constant pressure is determined by the entropy, and Eq. (40) shows that the pressure dependence of the Gibbs energy at constant temperature is determined by the volume.
Because dG is an exact differential, it follows directly that the second cross partial derivatives of Eq. (37) are equal, as given by Eq. (41).
This property is characteristic of an exact differential. Thus, from Eqs. (39), (40), and (41), the Maxwell relation, Eq. (42),
is obtained. This relation tells how the entropy changes with pressure at constant temperature.
Affinity and chemical equilibrium
Many chemical systems can be considered closed systems in which a single parameter ξ can be defined as a measure of the extent of the reaction or the degree of advancement of a process. If the reaction proceeds or the process advances spontaneously, then entropy must be produced according to the second law, and the uncompensated heat, defined by Eq. (43),
must be positive. In terms of the advancement parameter ξ, the uncompensated heat δa is given by Eq. (44),
where A is the affinity of the process or reaction. The affinity is related to internal entropy production diS by Eq. (45).
The condition that the entropy production is zero represents equilibrium, and hence A = 0 is an equivalent condition for equilibrium in a closed system. For spontaneous processes, because the signs of A and dξ must be the same, for positive A the process must advance or go in a forward direction in the usual sense of chemical reactions or physical processes, while for negative A the process must proceed in the reverse direction.
For a process that involves only pressure-volume work, Eqs. (17), (36), (43), and (44) can be combined to give Eqs. (46). These equations indicate that the affinity is a state function, given by Eq. (47).
It is useful to consider a closed system in which a chemical reaction can be characterized by the stoichiometry of reaction (48).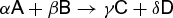 (48)
(48)
This stoichiometry requires that at each time element t in the reaction the number of moles of the ith component ni be given by Eq. (49),
where ni0 is the number of moles of i in the initial or original state (t = 0). The quantity υi is the stoichiometric coefficient for the ith component as given in the balanced equation (the convention is that υi is positive for products and negative for reactants), and ξ is the degree-of-advancement parameter, whose range is zero to unity. In terms of differential changes in advancement, Eq. (50)
holds.
For closed systems (constant temperature and pressure) in which only thermal, expansion, and chemical work terms are included, Eqs. (51) and (52)
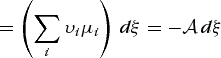 (51b)
(51b)hold. The condition for equilibrium is A = 0, and thus for a chemical reaction, equilibrium is achieved when Eq. (53) holds. If electrical work is included in Eq. (51), Eqs. (54) and (55)
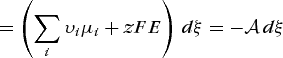 (54b)
(54b)hold, where z is the number of units of charge and F is the Faraday constant (the magnitude of the charge per mole of electrons); F = eNA, where e is the electron charge and NA is Avogadro's number. Because A = 0 is the equilibrium condition, equilibrium in an electrochemical system is given by Eq. (56).
More than one reaction can take place in a chemical system, each characterized by a degree-of-advancement parameter ξr, and thus for r independent reactions, Eqs. (57) and (58) hold, where the equilibrium condition is Eq. (59).
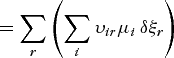 (57c)
(57c)At equilibrium, each of the affinities Ar must be zero; but for the spontaneous condition, inequality (60) holds. In a two-reaction system inequality (61)
holds, but now A1 and dξ1 do not necessarily have the same sign. If their signs are different, then the first reaction can be driven in the nonspontaneous direction by the second reaction. The reactions are then said to be coupled; reaction coupling is a common situation in biological systems. See also: Chemical equilibrium
Third law of thermodynamics
Although implicit in the concept of the existence of the fundamental equations of state for U or S is an absolute value of these functions, and therefore an extensive quantity which could be calculated on the basis of molecular properties from quantum mechanics and statistical mechanics, neither the first nor second law considers anything but differences in these state functions. The second law does suggest the existence of an absolute zero for an intensive variable, the temperature, but this is not sufficient to bridge the areas of classical and statistical thermodynamics. See also: Absolute zero
It is found experimentally, for many isothermal processes involving pure phases, that Eq. (62)
applies. This equation includes phase transitions between different crystalline modifications, solid-state chemical reactions, and even the solid-liquid and liquid-liquid isotope transitions in helium. This equation along with Eq. (63),
obtained from Eq. (31), implies that at zero absolute temperature the entropy of pure crystalline phases of the same substance or different substances are equal. If the entropies of pure phase are equal at T = 0, then it is reasonable to take the value of S(0) to be zero. A statement of the third law then is that the entropy of all pure crystalline fully ordered phases at T = 0 is zero. This makes it possible, by using Eq. (63), to calculate the absolute or third-law entropy of a substance from experimental measurements of its heat capacities. Comparison of such experimental values with those calculated by statistical thermodynamic methods has provided evidence for the validity of the third law. In some cases, thorough investigation of apparent discrepancies from the third law has led to new conclusions concerning the molecular structure of the substances or new information on the energy level system for the molecules. Calculation of the thermodynamic properties for a gas from the spectroscopic properties of molecules is an important result stemming from the third law. An alternative statement of the third law is that the absolute zero of temperature is unattainable in any real process. See also: Statistical mechanics
Thermodynamics of irreversible processes
From a mathematical standpoint, if the equations describing a time-dependent physical process change sign when the time variable t is changed to −t, then the process is said to be irreversible. If the substitution of t by −t does not change the equations, then the process described is said to be reversible. An example of an irreversible thermodynamic process is the approach to thermal equilibrium, which is described by the Fourier equation (64),
where T is the temperature, t is the time, and α is a parameter called the thermal diffusivity. The physicochemical processes of diffusion, heat and electrical conduction, and chemical reactions are irreversible processes. See also: Conduction (electricity); Conduction (heat); Diffusion
By utilizing Eqs. (44) and (26), the entropy production is found to be given by Eq. (65),
where A, the affinity of the chemical reaction, is related to the chemical potentials by Eq. (52). The reaction rate v is defined by Eq. (66), and thus Eq. (67)
holds. It is clear that A and v always have the same sign and that the affinity A is directly related to entropy production.
If several reactions are involved simultaneously, then Eq. (68)
holds, where the affinities are related to the chemical potentials by Eq. (58). At equilibrium all of the affinities are zero. The entropy production per unit time is given by Eq. (69).
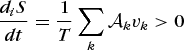 (69)
(69)The net entropy production arising from the simultaneous reactions must be positive. However, it is possible to have the inequalities (70) in a two-reaction system, provided that inequality (71)
is satisfied. As noted above, such reactions are said to be coupled in that a reaction can proceed in a direction opposed to its affinity, a phenomenon frequently encountered in biological systems. For example, matter may diffuse against its concentration gradient with negative entropy production driven by positive entropy production arising from heat flow.
Equation (69) can be rewritten as Eq. (72), where Jk and Xk are defined by Eqs. (73).
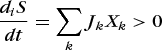 (72)
(72)The variable Jk is called a flux (rate) and the variable Xk is called a (generalized) force. At thermodynamic equilibrium Eqs. (74)
are satisfied for all values of k.
Close to equilibrium, various empirical laws, such as Fourier's law for heat flow and Fick's law for diffusion, show a linear relation between the Jk and Xk values. Such linear laws are called phenomenological relations. Although such relations do not necessarily hold far from equilibrium, it will be assumed that they do hold close to equilibrium.
It is useful to consider the case of two simultaneous irreversible processes which, in general, satisfy Eqs. (75).
The coefficients Lij are called phenomenological coupling coefficients. For example, L11 and L22 may denote heat conductivity and diffusion, while L12 describes the interference between these two processes (that is, thermodiffusion). Thus the Lij (i ≠ j) are called interference coupling coefficients.
Substitution of Eq. (75) into Eq. (72) yields Eq. (76),
where the equality sign prevails when X1 = X2 = 0. From algebra it follows that the Lij values must satisfy inequalities (77) and (78).
Thus while L11 and L22 must be positive, L12 and L21 may be positive or negative.
Onsager's theorem states that the coupling coefficients satisfy Eq. (79),
which is called the Onsager reciprocity relation after its derivation by Lars Onsager. These relations express the fact that if the flux Ji corresponding to the irreversible process i is coupled to the force Xj of the irreversible process j, then the flux Jj also is influenced by the force Xi through the same interference coefficient Lij. See also: Open-systems thermodynamics (biology); Osmoregulatory mechanisms; Thermoelectricity; Thermomagnetic effects
In the general case, Eq. (80)
holds, and when Lij ≠ 0 there is a coupling between the flux Ji and the force (gradient) Xj. The principle of microscopic reversibility implies that at least close to equilibrium Lij = Lji. Far from equilibrium the situation becomes much more complex, and a detailed molecular-level kinetic (as opposed to a phenomenological) approach is required in the analysis of such cases. See also: Thermodynamic principles; Thermodynamic processes

