
Key Concepts
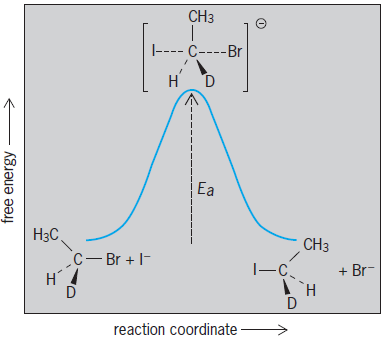
A complete, step-by-step account of how a reaction of organic compounds takes place. A fully detailed mechanism would correlate the original structure of the reactants with the final structure of the products and would account for changes in structure and energy throughout the progress of the reaction. A complete mechanism would also account for the formation of any intermediates and the rates of interconversions of all of the various species. Because it is not possible to detect directly all of these details of a reaction, evidence for a reaction mechanism is always indirect. Experiments are designed to produce results that provide logical evidence for (but can never unequivocally prove) a mechanism. For most organic reactions, there are mechanisms that are considered to be well established (that is, plausible) based on bodies of experimental evidence. Nevertheless, new data often become available that provide further insight into new details of a mechanism or that occasionally require a complete revision of an accepted mechanism. A common method for illustrating the progress of a reaction is the potential energy diagram, in which the free energy of the system is plotted as a function of the completion of the reaction (see illustration).
Classification of organic reactions
The description of an organic reaction mechanism typically includes designation of the overall reaction (for example, substitution, addition, elimination, oxidation, reduction, or rearrangement), the presence of any reactive intermediates (that is, carbocations, carbanions, free radicals, radical ions, carbenes, or excited states), the nature of the reagent that initiates the reaction (such as electrophilic or nucleophilic), the presence of any catalysis (such as acid or base), and any specific stereochemistry. For example, reaction (1)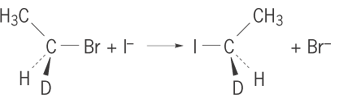 (1)
(1)
would be described as a concerted nucleophilic substitution of an alkyl halide that proceeds with inversion of stereochemistry. A reaction that proceeds in a single step, without intermediates, is described as concerted or synchronous. Reaction (1) is an example of the SN 2 mechanism (substitution, nucleophilic, bimolecular).
Potential energy diagrams
In a potential energy diagram (see illustration), the reaction coordinate is intended to represent the progress of the reaction, and it may or may not correlate with an easily observed or measurable feature. In reaction (1), the reaction coordinate could be considered to be the increasing bond length of the carbon-bromine (C-Br) bond as it is broken, or the decreasing separation of C and iodine (I) as they come together to form a bond. In fact, a complete potential energy diagram should illustrate the variation in energy as a function of both of these (and perhaps several other relevant structural features), but this would require a three-dimensional (or higher) plot.
Besides identifying the energy levels of the original reactants and the final products, the potential energy diagram indicates the energy level of the highest point along the reaction pathway, called the transition state. It is important to recognize that the reaction pathway actually taken in a reaction mechanism is chosen because it represents the lowest-energy pathway. The analogy often used is a mountain pass between two valleys, in which the top of the pass represents the transition state; the top of the pass represents the highest point traveled, but not the highest point in the vicinity. Because the transition state represents the highest energy that the molecules must attain as they proceed along the reaction pathway, the energy level of the transition state is a key indication of how easily the reaction can occur. Features that tend to make the transition state more stable (lower in energy) make the reaction more favorable. Such stabilizing features could be intramolecular, such as electron donation or withdrawal by substituents, or intermolecular, such as stabilization by solvent. See also: Chemical bonding; Energy
Kinetics
Another way to illustrate the various steps involved in a reaction mechanism is as a kinetic scheme that shows all of the individual steps and their rate constants. The SN 2 mechanism is a single step, so the kinetics must represent that step; the rate is observed to depend on the concentrations of both the organic substrate and the nucleophile. However, for multistep mechanisms the kinetics can be a powerful tool for distinguishing the presence of alternative pathways. For example, when more highly substituted alkyl halides undergo nucleophilic substitution, the rate is independent of the concentration of the nucleophile. This evidence suggests a two-step mechanism, called the SN1 mechanism, as shown in reaction scheme (2), where the k terms represent rate constants.
The SN
1 mechanism accomplishes the same overall nucleophilic substitution of an alkyl halide, but does so by initial dissociation of the leaving group (Br−) to form a carbocation, step (2a
). The nucleophile then attaches to the carbocation to form the final product, step (2b
).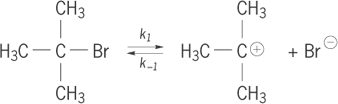 (2a)
(2a)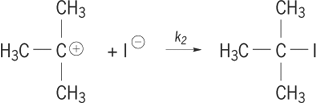 (2b)
(2b)
Alkyl halides that have bulky groups around the carbon to be substituted are less likely to be substituted by the direct SN2 mechanism, because the nucleophile encounters difficulty in making the bond to the inaccessible site (called steric hindrance). If those alkyl groups have substituents that can support a carbocation structure, generally by electron donation, then the SN1 mechanism becomes preferable. A crucial feature of a multistep reaction mechanism is the identification of the rate-determining step. The overall rate of reaction can be no faster than its slowest step. In the SN1 mechanism, the bond-breaking reaction (2a ) is typically much slower than the bond-forming reaction (2b ). Hence, the observed rate is the rate of the first step only. Thus, kinetics can distinguish the SN 1 and SN 2 mechanisms, as shown in Eqs. (3) and (4),
where R is an alkyl group, X is a halogen or other leaving group, Nu is a nucleophile, and the terms in the brackets represent concentrations.
A more complete description of the SN1 mechanism was recognized when it was observed that the presence of excess leaving group [for example, Br− in reaction (2)] can affect the rate (called the common ion rate depression). This indicated that the mechanism should include a reverse step [k−1 in reaction (2a)] in which the leaving group returns to the cation, regenerating starting material. In this case, the rate depends in a complex manner on the competition of nucleophile and leaving group for reaction with the carbocation. See also: Chemical dynamics; Reactive intermediates; Steric effect (chemistry)
Activation parameters
The temperature dependence of the rate constant provides significant information about the transition state of the rate-determining step. The Arrhenius equation (5)
expresses that dependence in terms of an exponential function of temperature and an activation energy, Ea; A is called the Arrhenius or preexponential factor, and R is the gas constant. See also: Gas; Logarithm
The activation energy represents the energy difference between the reactants and the transition state; that is, the amount of energy that must be provided in order to proceed along the reaction pathway successfully from reactant to product (see illustration).
A more complete description of transition-state theory expresses the rate constant in terms of contributions from both an enthalpy of activation, ΔH‡, and an entropy of activation, ΔS‡. In this case, enthalpy of activation represents the additional enthalpy content of the transition state compared to reactants; ΔH‡ can be rather directly correlated with the energy of activation. The entropy of activation, ΔS‡, represents the additional entropy content of the transition state compared to reactants; ΔS‡ is highly informative with respect to the reaction mechanism, because it indicates whether the overall structure becomes more ordered or more disordered as the reaction proceeds from reactants to transition state. For example, the transition state for the SN2 reaction (see illustration) requires the reactants to be organized in a very specific manner, with the nucleophile approaching the opposite side of the leaving group; the SN2 reaction typically shows a negative value for ΔS‡. See also: Enthalpy; Entropy
Stereochemistry
Careful attention to the stereochemistry of a reaction often provides crucial insight into the specific orientation of the molecules as they proceed through the reaction mechanism. The complete inversion of stereochemistry observed in the SN2 mechanism provides evidence for the backside attack of the nucleophile. Alkyl halides that undergo substitution by the SN1 mechanism do not show specific stereochemistry, since the loss of the leaving group is completely uncorrelated with the bonding of the nucleophile.
In addition reactions, possible stereochemical outcomes are addition of the new bonds to the same or opposite sides of the original pi bond, called syn and anti addition, respectively. The anti addition of bromine to double bonds provides evidence for the intermediacy of a bridged bromonium ion, as shown in reaction (6).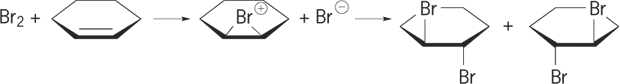 (6)
(6)
Experimental probes of mechanisms
Chemists use a variety of techniques to determine the mechanistic course of reactions. The most direct is actual observation of the reactants, intermediates, and products in real time during the reaction. Fast spectroscopic techniques are becoming ever faster and more sensitive, so that direct evidence for short-lived intermediates is often obtainable. A representative fast technique is flash photolysis, in which a compound is rapidly decomposed by an intense pulse of light. Laser light pulses less than a picosecond (10−12 s) are available; they, combined with detection methods that can record data on a time scale of picoseconds, allow for direct observation of the spectroscopic or other properties of reaction intermediates. See also: Laser spectroscopy; Ultrafast molecular processes
Another approach is to create an environment that is stabilizing for the reaction intermediates to allow study of their properties by standard techniques. A common approach called matrix isolation creates the intermediate in a frozen matrix, so that it is unable to diffuse and react with other species. In this case, the reactive intermediate must be formed by a unimolecular reaction, since it cannot diffuse to meet another reactant, and typically light-induced photodecompositions are used. An example of the successful use of matrix isolation to generate and observe benzyne is shown in reaction (7).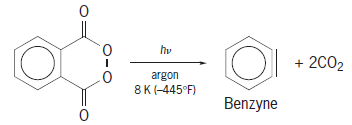 (7)
(7)
Isolating benzyne and collecting spectroscopic evidence for its structure under matrix isolation conditions provides evidence that it could be involved as an intermediate in the same reaction under other conditions. At room temperature in solution, benzyne reacts with a variety of substrates to form cycloaddition products, such as in reaction (8).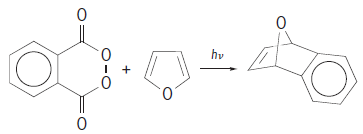 (8)
(8)
The observation of these same products upon warming a frozen matrix containing benzyne plus the added substrate helps to verify its reactivity patterns and strengthens its implication as a reaction intermediate. See also: Matrix isolation
Substituent effects and Hammett equation
By far the major methods for determining mechanisms of reactions as they occur under normal laboratory conditions utilize kinetics and stereochemistry, as already illustrated for the differentiation of SN1 and SN2 substitution mechanisms. Variations in the observed kinetics as the structure of the reactants are systematically varied are the basis of one of the most powerful methods of detecting details of a reaction mechanism. Correlation of substituent effects by the Hammett equation is a standard approach that allows the prediction of the extent of electron shifting in a reaction mechanism. Substituents are assigned substituent constants (σ) that indicate the degree to which they are electron-donating (negative values) or electron-withdrawing (positive values) in a standard reaction (the dissociation of benzoic acid). Reactions then display a dependence on the substituent constants, a reaction constant (ρ) that indicates the degree to which the reaction is favored by electron donation (negative values) or favored by electron withdrawal (positive values). The negative ρ value indicates that the reaction is favored by substituents that donate electron density, a point that would not have been obvious from looking at the reactants and products, all of which are uncharged. This evidence suggests that the transition state is polarized. In that transition state, electron donation provides a stabilizing effect, lowers the energy of the transition state, and thereby increases the rate. See also: Substitution reaction
Isotope effects and isotopic labeling
The use of isotopes is an important tool in mechanistic studies. Kinetic isotope effects are often subtle but useful methods to distinguish details of the transition state. The magnitude of the kinetic isotope effect provides a measure of the amount of bond breaking at the transition state. Isotopes are also used simply as labels to distinguish otherwise identical atoms. For example, the use of 13C isotopic labeling in reaction (9),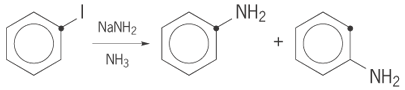 (9)
(9)
where the heavy dot represents the position of a 13C atom, provided evidence that the mechanism was not a simple nucleophilic substitution. The appearance of 13C equally to two different positions of the product is evidence for a symmetrical intermediate such as benzyne. See also: Deuterium; Isotope
Theoretical correlations
The theory underlying organic chemistry has developed to the stage that both qualitative and quantitative approaches often provide excellent insight into the workings of a reaction mechanism. The principle of conservation of orbital symmetry, developed by R. B. Woodward and R. Hoffmann and often called the Woodward-Hoffmann rules, provides a simple yet powerful method for predicting the stereochemistry of concerted reactions. The principle states that the formation of new bonds and the breaking of old bonds will be carried out preferentially in a manner that maximizes bonding at all times. See also: Pericyclic reaction; Woodward-Hoffmann rule
By knowing the symmetry properties of the molecular orbitals of reactants and products, the preferred (or allowed) pathway can be predicted. The approach can be greatly simplified by recognizing the cyclic delocalized transition state of the reaction and counting the number of electrons involved in the cyclic transition state. If there are (4n + 2) electrons (that is, 2, 6, 10, … electrons), the reaction is considered favorable, because all of the bonding can occur on the same sides of the molecules. Thus cycloadditions such as the Diels-Alder reaction (10)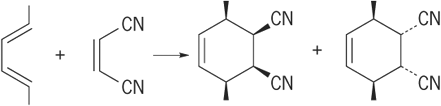 (10)
(10)
are favorable according to the Woodward-Hoffmann rules, because they involve six electrons in the delocalized transition state.
Reactions involving 4n electrons, such as the cycloaddition of two double bonds to form a four-membered ring, are unfavorable by these rules and are not observed except in photochemical reactions, for which the Woodward-Hoffmann rules are exactly reversed. See also: Delocalization; Diels-Alder reaction; Molecular orbital theory; Photochemistry
The improvement of computing capabilities has allowed quantitative calculations to become accurate enough to predict energies and structures for simple molecules. Quantum-mechanics calculations may be performed either from first principles, that is, without simplifying assumptions, or semiempirically, that is, with some standardizing parameters. The desired result is a potential energy map that correlates the energy levels of the possible structures of the reactants as they transform to products. Such approaches can help to rule out possible pathways as too high in energy or can suggest alternative pathways that appear feasible based on calculated energies. See also: Computational chemistry; Molecular mechanics; Quantum chemistry

