
Key Concepts
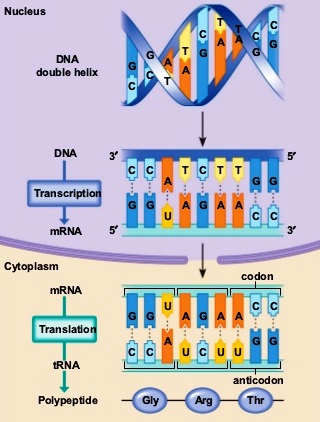
The basic unit in inheritance consisting of DNA. A gene is composed of a deoxyribonucleic acid (DNA) sequence that contains the elements required for transcription of a complementary ribonucleic acid (RNA), which is sometimes the functional gene product, but more often is converted into messenger RNAs (mRNAs) that specify the amino acid sequence of a protein product (Fig. 1). The facts of Mendelian inheritance indicate the presence of discrete hereditary units (genes) that replicate at each cell division, producing remarkably exact copies of themselves, and that in some highly specific way determine the characteristics of the individuals that bear them. The evidence also shows that each of these units may at times mutate to give a new equally stable unit (called an allele), which has more or less similar, but not identical, effects on the characters of its bearers. The criteria for the recognition that certain genes are alleles have been that they (1) arise from one another by a single mutation, (2) have similar effects on the characters of the organism, and (3) occupy the same locus (region) in the chromosome. It has long been known that there were a few cases in which these criteria did not give consistent results, but these were explained by special hypotheses in the individual cases. However, such cases have been found to be so numerous that they appear to be the rule rather than the exception. See also: Allele; Cell division; Chromosome; Deoxyribonucleic acid (DNA); Mendelism; Mutation; Protein; Recombination (genetics); Ribonucleic acid (RNA); Transcription
The term gene, or cistron, also may be used to indicate a unit of function. Specifically, a cistron is used to designate a genetic unit (DNA fragment) that codes for a particular polypeptide. See also: Genetics; Peptide
Molecular biology
Every gene consists of a linear sequence of bases in a nucleic acid molecule. Genes are specified by the sequence of bases in DNA in prokaryotic, archaeal, and eukaryotic cells, and in DNA or RNA in prokaryotic or eukaryotic viruses. The flow of genetic information from DNA to mRNA to protein is historically referred to as the central dogma of molecular biology; however, this view required modification with the discovery of retroviruses, whose genetic flow goes from RNA to DNA by reverse transcription and then to mRNA and proteins. The ultimate expressions of gene function are the formation of structural and regulatory RNA molecules and proteins. These macromolecules carry out the biochemical reactions of cells and provide the structural elements that make up cells. See also: Molecular biology; Nucleic acid; Retrovirus
Flow of genetic information
One goal of molecular biology as it applies to genes is to understand the function, expression, and regulation of a gene in terms of its DNA or RNA sequence. The genetic information in genes that encode proteins is first transcribed from one strand of DNA into a complementary mRNA molecule by the action of the RNA polymerase enzyme. The genetic code is nearly universal for all prokaryotic, archaeal, and eukaryotic organisms. Many kinds of eukaryotic mRNA molecules and a limited number of prokaryotic mRNA molecules are further processed by splicing, which removes intervening sequences called introns. In some eukaryotic mRNA molecules, certain bases are also changed posttranscriptionally by a process called RNA editing. The genetic code in the resulting mRNA molecules is translated into proteins with specific amino acid sequences by the action of the translation apparatus, consisting of transfer RNA (tRNA) molecules, ribosomes, and many other proteins. The genetic code in an mRNA molecule is the correspondence of three contiguous (triplet) bases, called a codon, to the common amino acids and translation stop signals (Fig. 2); the bases are adenine (A), uracil (U), guanine (G), and cytosine (C). There are 61 codons that specify the 20 common amino acids, and 3 codons that lead to translation stopping; hence, the genetic code is degenerate because certain amino acids are specified by more than one codon. See also: Amino acid; Genetic code; Intron; Ribosomes
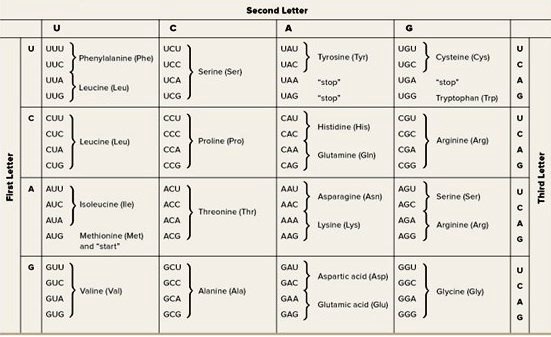
The sequence of amino acids in a protein is determined by the series of codons starting from a fixed translation initiation codon. AUG and GUG are the major translation start codons of prokaryotic genes. AUG is almost always the translation start codon of eukaryotic genes. The bacterial start AUG and GUG codons specify a modified form of methionine, whereas AUG or GUG codons internal to reading frames specify methionine or valine, respectively.
Isolating genes
In many cases, only genes that mediate a specific cellular or viral function are isolated. The recombinant DNA methods used to isolate a gene vary widely, depending on the experimental system, and genes from RNA genomes must be converted into a corresponding DNA molecule by biochemical manipulation using the enzyme reverse transcriptase. The isolation of the gene is referred to as cloning and allows large quantities of DNA corresponding to a gene of interest to be isolated and manipulated. See also: Cloning; Genetic engineering; Reverse transcriptase
After the gene is isolated, the sequence of the nucleotide bases can be determined. The goal of the large-scale Human Genome Project is to sequence all the genes of several model organisms (Fig. 3) and humans. The sequence of the region containing the gene can reveal numerous features. If a gene is thought to encode a protein molecule, the genetic code can be applied to the sequence of bases determined from the cloned DNA. The application of the genetic code is done automatically by computer programs, which can identify the sequence of contiguous amino acids of the protein molecule encoded by the gene. If the function of a gene is unknown, comparisons of its nucleic acid or predicted amino acid sequence with the contents of huge international databases can often identify genes or proteins with analogous or related functions. These databases contain all the known sequences from many prokaryotic, archaeal, and eukaryotic organisms. Putative regulatory and transcript-processing sites also can be identified by computer. These putative sites, called consensus sequences, have been shown to play roles in the regulation and expression of groups of prokaryotic, archaeal, or eukaryotic genes. However, computer predictions are just a guide and are not a substitute for analyzing expression and regulation by direct experimentation. See also: Genetic mapping; Human genome


