
Key Concepts
A scientific discipline concerned with genetically determined resemblances and differences among human beings. Human genetics analyzes all aspects of inheritance occurring in humans. Although the mechanism by which heredity operates remained mostly unknown until the twentieth century, the idea that certain physical and mental characteristics, normal or abnormal, can "run in families" goes back to ancient times. Formerly, genetics was thought to be concerned only with the familial transmission of rare and insignificant characteristics, but its fundamental biological role is now apparent. Genes, the units of heredity, have two unique properties: they are self-replicating, and they carry (in their biochemical structure) the codes for protein synthesis (Fig. 1). Consequently, genes play the double role of transmitting genetic information from generation to generation and of governing all the activities of living cells. See also: Biochemistry; Gene; Genetics; Human genome; Protein
Expansion of the knowledge of human genetics has come from various directions. For example, technological advances in the visualization of human chromosomes have shown that abnormalities of chromosome number or structure are surprisingly common and of many different kinds, and that they account for birth defects or mental impairment in many individuals, as well as for numerous early spontaneous abortions. Progress in molecular biology has clarified the molecular structure of chromosomes and their constituent genes, the mechanisms of deoxyribonucleic acid (DNA) synthesis and protein synthesis, and the ways in which change in the molecular structure of a gene can lead to a disease. In addition, research in human genetics has been stimulated by concerns about possible genetic damage through environmental agents, particularly ionizing radiation, and the possible harmful effects of hazardous substances in the environment on prenatal development. See also: Deoxyribonucleic acid (DNA); Disease; Environmental toxicology; Radiation injury to plants and animals
Chromosome and gene structure
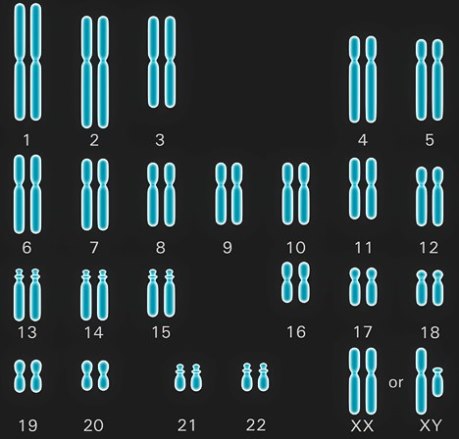
In normal humans, the nucleus of each normal cell contains 46 chromosomes, which comprise 23 different pairs (Fig. 2). Of each chromosome pair, one is paternal in origin and the other is maternal in origin. In turn, only one member of each pair is handed on through the reproductive cell (egg or sperm) to each child. Thus, each egg or sperm has only 23 chromosomes, the haploid number; the fusion of egg and sperm at fertilization will restore the double, or diploid, chromosome number of 46. See also: Chromosome; Fertilization (animal)
The segregation of chromosome pairs during meiosis allows for a large amount of “shuffling” of genetic material as it is passed down the generation. With 23 pairs of chromosomes, the total possible number of different chromosome combinations in the gametes is 223, or about 8 million. Two parents can provide 223 × 223 different chromosome combinations. This enormous source of variation is multiplied still further by the mechanism of crossing over, in which homologous chromosomes exchange segments during meiosis. See also: Crossing-over (genetics); Meiosis
Twenty-two of the 23 chromosome pairs, the autosomes, are alike in both sexes; the other pair comprises the sex chromosomes (Fig. 2). A female has a pair of X chromosomes; a male has a single X, which is paired with a Y chromosome that he has inherited from his father and will transmit to each of his sons. Sex is determined at fertilization and depends on whether the egg (which has a single X chromosome) is fertilized by an X-bearing or a Y-bearing sperm. See also: Sex determination
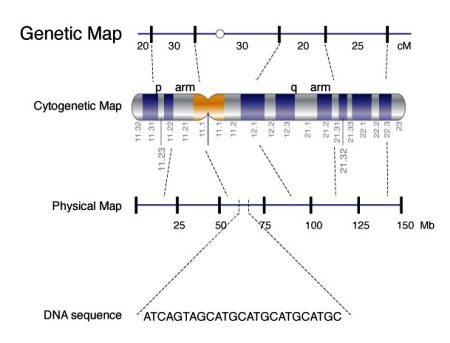
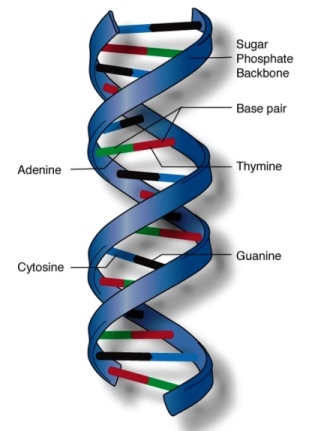
All chromosomes are composed of DNA (Fig. 3), and genes are segments of DNA. There are about 19,000–20,000 genes on the 46 human chromosomes, and many have been mapped to specific chromosomal locations (Fig. 1). The genetic information is coded in DNA in the form of triplets of four bases: two purines, adenine (A) and guanine (G), and two pyrimidines, thymine (T) and cytosine (C). Each triplet combination (codon) codes for a specific amino acid. The sequence of bases in a specific gene dictates the sequence of amino acids in the specific protein coded by that gene. See also: Amino acid; Genetic code; Genetic mapping
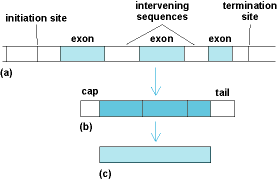
A gene initiates synthesis of a protein by transcription of the DNA into messenger ribonucleic acid (mRNA), which is a single-stranded molecule complementary to the DNA. After processing within the cell nucleus, the mRNA moves into the cytoplasm, where it binds to ribosomes. The translation of RNA into protein takes place on the ribosomes. In addition, a typical gene is not a simple uninterrupted length of DNA. Most genes are made up of coding sequences (exons) separated by noncoding regions (intervening sequences, or introns). Transcription of the gene into RNA begins at an initiation site in advance of the first coding sequence and terminates beyond the end of the last sequence. After the entire DNA segment has been transcribed into mRNA, the intervening sequences are removed, and the coding sequences are spliced together before the mRNA is translated into a polypeptide (Fig. 4). See also: Exon; Intron; Ribonucleic acid (RNA); Ribosomes
Any gene occupies a specific chromosomal position, or locus. The alternative genes at a particular locus are said to be alleles. If a pair of alleles are identical, the individual is homozygous; if they are different, the individual is heterozygous. See also: Allele
Mutation
Genetic variation has its origin in mutation. Although any change in DNA is a mutation (whether it is a microscopically detectable change in the structure of a chromosome or a single base change in the genetic code), the term is usually applied to a stable change in DNA that alters the genetic code and thus leads to synthesis of an altered protein. Mutation can occur in reproductive cells or somatic cells, but the genetically significant ones are those that occur in reproductive cells and can be transmitted to future generations. Natural selection acts upon the genetic diversity generated by mutation to preserve beneficial mutations and eliminate deleterious ones. See also: Mutation
A very large amount of genetic variation exists in the human population. Everyone carries many mutations; some are newly acquired, but others are inherited through innumerable generations. Although the exact number is unknown, it is likely that everyone is heterozygous at numerous loci, perhaps as many as 20%. See also: Population genetics
Single-gene inheritance
The patterns of inheritance of characteristics determined by single genes or gene pairs depend on two conditions: (1) whether the gene in question is on an autosome (autosomal inheritance) or on the X chromosome (X-linked inheritance); and (2) whether the gene is dominant (expressed in heterozygotes, when it is present on only one member of a chromosomal pair and has a normal allele), or is recessive (expressed only in homozygotes, when it is present on both chromosomes). See also: Dominance
Quantitative inheritance
A quantitative trait is one that is under the control of many factors, both genetic and environmental, with each contributing only a small amount to the total variability of the trait. The phenotype may show continuous variation (for example, height and skin color) or quasicontinuous variation (taking only integer values, such as the number of ridges in a fingerprint), or it may be discontinuous [a presence/absence trait, such as diabetes or intellectual disability (formerly termed mental retardation)]. With discontinuous traits, it is assumed that there exists an underlying continuous variable and that individuals having a value of this variable above (or below) a threshold possess the trait.
A trait that “runs in families” is said to be familial. However, not all familial traits are hereditary because relatives tend to share common environments as well as common genes. It is the major task of quantitative genetics to disentangle the effects of environment and heredity, but this task is not easy in humans because the two factors are often confounded. A rough measure of the relative importance of heredity and environment is an index called heritability. The total variance (a statistical measure of variability) of a trait can be partitioned into a genetic variance and an environmental variance, at least in simple cases. The quantitative model can be expressed as
Heritability is then defined as
For example, the heritability of height in humans is about 0.75; that is, about 75% of the total variance in height is due to variability in genes that affect height and 25% is due to exposure to different environments.
Hereditary diseases
Medical genetics (that is, genetic counseling, including prenatal diagnostics) has become an integral part of preventive medicine. It has contributed increasingly to systematics of disease, diagnostics, and even therapy. Because many external causes of disease, including infections, have been brought under control in today's modern world, doctors can devote much of their skill to treating diseases from internal sources, that is, hereditary diseases, or those brought about by the interaction of genetic predispositions with certain stress factors in the environment. Hereditary diseases may be subdivided into three classes: chromosomal diseases; hereditary diseases with simple, mendelian modes of inheritance; and multifactorial diseases. See also: Medicine; Prenatal diagnosis
Chromosomal diseases
One of every 200 newborns suffers from an abnormality that is caused by a microscopically visible deviation in the number or structure of chromosomes. Such chromosomal aberrations are much more common (approximately 40%) among spontaneous miscarriages. The most important clinical abnormality among the survivors is Down syndrome—a condition due to trisomy of chromosome 21, one of the smallest human chromosomes. The term trisomy means that this chromosome is present three times (instead of twice); the entire chromosome complement therefore comprises 47 (not 46) chromosomes. Down syndrome occurs one to two times in every 1000 births, and it is a good example of a characteristic pattern of abnormalities that is produced by a single genetic defect. See also: Chromosome aberration; Down syndrome
Other autosomal aberrations observed in living newborns that lead to characteristic syndromes include trisomies 13 and 18 (both very rare), as well as a variety of structural aberrations, including translocations (exchanges of chromosomal segments among different chromosomes) and deletions (losses of chromosome segments). Translocations normally have no influence on the health status of the individual if there is no gain or loss of chromosomal material (these are called balanced translocations). However, carriers of balanced translocations usually run a high risk of having “unbalanced” offspring, that is, children in whom the same translocation causes the gain or loss of genetic material and who suffer from a characteristic malformation syndrome.
Clinical syndromes caused by specific aberrations vary, but certain clinical signs are common: low birth weights; a peculiar face; delayed general, and especially mental, development, often leading to severe mental deficiency; and multiple malformations, including the abnormal development of limbs, heart, and kidneys. See also: Congenital anomalies
Less severe signs than those caused by autosomal aberrations are found in individuals with abnormalities in the number (and structure) of sex chromosomes. This is because, in individuals having more than one X chromosome, the additional X chromosomes are inactivated early in pregnancy. For example, in women, one of the two X chromosomes is always inactivated. Inactivation occurs at random so that every normal woman is a mosaic of cells in which either one or the other X chromosome is active. Additional X chromosomes that an individual may have received will also be inactivated; thus, genetic imbalance is avoided to a certain degree. However, inactivation is not complete; therefore, individuals with trisomies [for example, XXY (Klinefelter syndrome), XXX (triple-X syndrome), or XYY] or monosomies (XO; Turner syndrome) often show abnormal sexual development, intelligence, or behavior.
Diseases with mendelian inheritance
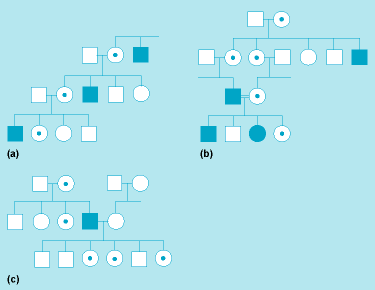
In contrast to chromosomal aberrations, the genetic defects in hereditary diseases with simple, mendelian modes of inheritance cannot be recognized by microscopic examination; as a rule, they must be inferred more indirectly from the phenotype and the pattern of inheritance in pedigrees (Fig. 5). The defects are found in the molecular structure of the DNA. Often, one base pair only is altered, although sometimes more complex molecular changes (such as deletions of some bases or abnormal recombination) are involved. Approximately 1% of all newborns have, or will develop during their lives, a hereditary disease showing a simple, mendelian mode of inheritance. See also: Mendelism
In medical genetics, a condition is called dominant if the heterozygotes deviate in a clearly recognizable way from the normal homozygotes, in most cases by showing an abnormality. Because such dominant mutations are usually rare, almost no homozygotes are observed and their clinical condition is typically unknown. In exceptional instances, homozygotes for dominant conditions have been described; they have usually shown more severe clinical signs than the heterozygotes.
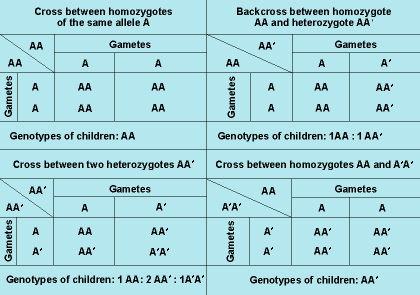
In Fig. 6, there are four mendelian crosses for one pair of alleles, A and A′. Cross no. 2, the backcross between the normal homozygote and the heterozygote, which leads to 1:1 segregation, is found most commonly in autosomal-dominant inheritance. Crosses 3 and 4 are extremely rare because homozygotes for an abnormal allele are usually rare. In autosomal-recessive inheritance, backcrosses between normal homozygotes and heterozygotes (cross no. 2) are also common; however, because heterozygotes are normal phenotypically, such crosses will go unnoticed in most instances. The most common cross leading to homozygous, clinically affected offspring is the intercross between two heterozygotes, AA′, which results in 1:2:1 segregation.
The pattern of inheritance is often determined by studying the history of a trait among a group of relatives. As an example, a pedigree in which a rare autosomal-dominant condition is transmitted through four generations is shown in Fig. 7. The mutant allele is located on an autosome; the affected individuals are heterozygous. Because one of the two alleles at this gene locus is altered by mutations and each of the two alleles has a 50% probability of being transmitted to a child (Fig. 6, cross no. 2), each child has a 50% risk of being affected. Because this mutation is autosomal, it is transmitted independently of the sex chromosomes, and the risk of being affected is not influenced by the sex of the parents or child.
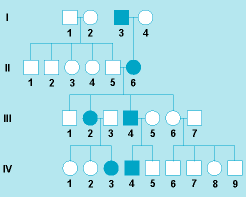
In some dominant conditions, the harmful phenotype may not be expressed in a gene carrier (this is called incomplete penetrance), or clinical signs may vary in severeness among carriers (this is called variable expressivity). Penetrance and expressivity may be influenced by other genetic factors, including the sex of the affected person; in other instances, the constitution of the “normal” allele has been implicated. Environmental conditions may occasionally be important. In most cases, however, the reasons are unknown.
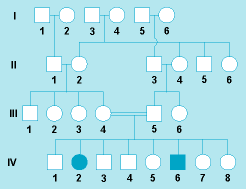
Inheritance also can occur in an autosomal-recessive manner. A pedigree for albinism, that is, with an autosomal-recessive trait, is shown in Fig. 8. The affected individuals (IV, 2 and 6) have two mutant alleles (one from each parent), and they are homozygous for the albinism gene. Hence, their unaffected parents (III, 4 and 5) must both be heterozygous for this allele. The probability of two individuals having the same allele increases when some of their genes come from a common ancestor (that is, when they are relatives). An increase of matings between relatives (for example, first cousins) in comparison with the population average is characteristic for rare autosomal-recessive diseases.
Finally, X-linked modes of inheritance occur when the mutant allele is located on the X chromosome. The most important X-linked mode of inheritance is the recessive one (Fig. 9). Here, the males (referred to as hemizygotes, having only one allele) are affected because they have no normal allele. The female heterozygotes, on the other hand, will be unaffected because the one normal allele is sufficient for maintaining function. A classical example is hemophilia A, in which one of the serum factors necessary for normal blood clotting is inactive or lacking. (The disease can now be controlled by repeated substitution of the deficient blood factor; this is a good example for phenotypic therapy of a hereditary disease by substitution of a deficient gene product.) As shown in Fig. 9a , male family members are affected, whereas their sisters and daughters, while being unaffected themselves, transmit the mutant gene to half their sons. Only in very rare instances, when a hemophilic patient marries a heterozygous carrier, are homozygous females observed (Fig. 9b). See also: Hemophilia; Sex-linked inheritance
Multifactorial diseases
There are thousands of hereditary diseases with simple, mendelian modes of inheritance, but most common anomalies and diseases are influenced by genetic variability at more than one gene locus. Most congenital malformations, including congenital heart disease, cleft lip and palate, and neural tube defects, fall into this category, as do the constitutional diseases, such as diabetes mellitus, coronary heart disease, anomalies of the immune response, and many mental diseases, including schizophrenia and affective disorders. All of these conditions are common and often increase in frequency with advanced age. Moreover, the environment usually contributes significantly to the disease risk.
Biochemical genetics
Research dealing with biochemical genetics began with the study of inborn errors of metabolism. These are diseases of the body chemistry in which a small molecule, such as a sugar or amino acid, accumulates in body fluids because an enzyme responsible for its metabolic breakdown is deficient. This molecular defect is the result of a mutation (or mutations) in the gene coding for the enzyme protein. The accumulated molecule, dependent on its nature, is responsible for the causation of a highly specific pattern of disease. In addition, similar heritable defective enzymes have been found to interfere with the breakdown of very large molecules, including mucopolysaccharides and the complex lipids that are such prominent components of brain substance. The resultant storage disorders present with extreme alterations in morphology and bony structure and with neurodegenerative disease. See also: Enzyme; Metabolic disorders
Inheritance mechanism
Most hereditary disorders of metabolism are inherited in an autosomal-recessive fashion. In these families, each parent carries a single mutant gene on one chromosome and a normal gene on the other. Typically, these mutations are rare. In populations where consanguinity is common, rare recessive diseases are seen with relative frequency, and affected individuals are homozygous for the same mutation. In populations with more genetic diversity, most affected individuals carry two different mutations in the same gene. Some metabolic diseases are coded for by genes on the X chromosome. Most of these disorders are fully recessive; thus, affected individuals are all males, whereas females carrying the gene are clinically normal. The disorders that result from mutations in the mitochondrial genome are inherited in nonmendelian fashion because mitochondrial DNA is inherited only from the mother. Those that carry a mutation are heteroplasmic; that is, each carries a mixed population of mitochondria, some with the mutation and some without the mutation.
Inborn errors of amino acid metabolism
Phenylketonuria (PKU) is a prototypic biochemical genetic disorder. It is an autosomally recessive disorder in which mutations demonstrated in a sizable number of families lead, when present in the genes on both chromosomes, to defective activity of the enzyme that catalyzes the first step in the metabolism of phenylalanine. This results in the accumulation of phenylalanine and a recognizable clinical disease whose most prominent feature is severe intellectual disability of mental development. See also: Intellectual disability; Phenylketonuria
Mitochondrial disease
The diseases that result from mutation in mitochondrial DNA have been recognized as such only since the 1990s. They result from point mutations, deletions, and other rearrangements. Many of the disorders are known as mitochondrial myopathies (diseases of muscles) because skeletal myopathy or cardiomyopathy are characteristic features. One notable disease resulting from mutation in mitochondrial DNA is known as mitochondrial encephalomyopathy, lactic acidosis, and strokelike episodes (MELAS). In general, most of the disorders express themselves chemically in elevated concentrations of lactic acid in the blood or cerebrospinal fluid. See also: Mitochondria

