
Key Concepts
Laws governing the transformation of energy. Thermodynamics is the science of the transformation of energy. It differs from the dynamics of English physicist and mathematician Isaac Newton by taking into account the concept of temperature, which is outside the scope of classical mechanics. In practice, thermodynamics is useful for assessing the efficiencies of heat engines (devices that transform heat into work, see Fig. 1) and refrigerators (devices that use external sources of work to transfer heat from a hot system to cooler sinks), and for discussing the spontaneity of chemical reactions (their tendency to occur naturally) and the work that they can be used to generate. See also: Chemistry; Energy; Heat; Newton's laws of motion; Physics; Refrigeration; Refrigerator; Temperature

The subject of thermodynamics is founded on four generalizations of experience, which are called the laws of thermodynamics. Each law embodies a particular constraint on the properties of the world. The connection between phenomenological thermodynamics and the properties of the constituent particles of a system is established by statistical thermodynamics, also called statistical mechanics. Classical thermodynamics consists of a collection of mathematical relations between observables, and as such is independent of any underlying model of matter (in terms, for instance, of atoms). However, interpretations in terms of the statistical behavior of large assemblies of particles greatly enriches the understanding of the relations established by thermodynamics, and a full description of nature should use explanations that move effortlessly between the two modes of discourse. See also: Atom; Matter; Statistical mechanics
There is a handful of very primitive concepts in thermodynamics. The first is the distinction between the system, which is the assembly of interest, and the surroundings, which is everything else. The surroundings are where observations on the system are carried out and attempts are made to infer its properties from those measurements. The system and the surroundings jointly constitute the universe and are distinguished by the boundary that separates them. If the boundary is impervious to the penetration of matter and energy, the system is classified as isolated. If energy but not matter can pass through it, the system is said to be closed. If both energy and matter can penetrate the boundary, the system is said to be open. Another primitive concept of thermodynamics is work. By "work," what is meant is a process by which a weight may be raised in the surroundings. Work is the link between mechanics and thermodynamics. See also: Work
None of these primitive concepts introduces the properties that are traditionally regarded as central to thermodynamics, namely temperature, energy, heat, and entropy. These concepts are introduced by the laws and are based on the foundations that these primitive concepts provide.
Zeroth law of thermodynamics
The zeroth law of thermodynamics establishes the existence of a property called temperature. This law is based on the observation that if a system A is in thermal equilibrium with a system B (that is, no change in the properties of B takes place when the two are in contact), and if system B is in thermal equilibrium with a system C, then it is invariably the case that A will be found to be in equilibrium with C if the two systems are placed in mutual contact. This law suggests that a numerical scale can be established for the common property, and if A, B, and C have the same numerical values of this property, then they will be in mutual thermal equilibrium if they were placed in contact. This property is now called the temperature.
In thermodynamics it is appropriate to report temperatures on a natural scale, where 0 is ascribed to the lowest attainable temperature. Temperatures on this thermodynamic temperature scale are denoted T and are commonly reported in kelvins. The relation between the Kelvin scale and the Celsius scale (θ) is T(K) = θ(°C) + 273.15. See also: Temperature
First law of thermodynamics
The first law of thermodynamics establishes the existence of a property called the internal energy of a system. It also brings into the discussion the concept of heat.
The first law is based on the observation that a change in the state of a system can be brought about by a variety of techniques. Indeed, if attention is confined to an adiabatic system, one that is thermally insulated from its surroundings, then the research of English physicist James Prescott Joule shows that same change of state is brought about by a given quantity of work regardless of the manner in which the work is done (Fig. 2). This observation suggests that, just as the height through which a mountaineer climbs can be calculated from the difference in altitudes regardless of the path the climber takes between two fixed points, so the work, w, can be calculated from the difference between the final and initial properties of a system. The relevant property is called the internal energy, U. However, if the transformation of the system is taken along a path that is not adiabatic, a different quantity of work may be required. The difference between the work of adiabatic change and the work of nonadiabatic change is called heat, q. In general, Eq. (1)
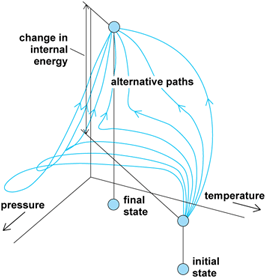
is satisfied, where ΔU is the change in internal energy between the final and initial states of the system. See also: Adiabatic process; Energy; Heat
The down-to-earth implication of this argument is that there are two modes of transferring energy between a system and its surroundings. One is by doing work; the other is by heating the system. Work and heat are modes of transferring energy. They are not forms of energy in their own right. Work is a mode of transfer that is equivalent (if not the case in actuality) to raising a weight in the surroundings. Heat is a mode of transfer that arises from a difference in temperature between the system and its surroundings. What is commonly called "heat" is more correctly called the thermal motion of the molecules of a system. See also: Molecule
The molecular interpretation of thermodynamics adds insight to the operational distinction between work and heat. Work is a transfer of energy that stimulates (or is caused by) organized molecular motion in the surroundings. Thus, the raising of a weight by a system corresponds to the organized, unidirectional motion of the atoms of the weight. In contrast, heat is the transfer of energy that stimulates (or is caused by) chaotic molecular motion in the surroundings. Thus, the emergence of energy as heat into the surroundings is the chaotic, tumbling-out of stored energy.
The first law of thermodynamics states that the internal energy of an isolated system is conserved. That is, for a system to which no energy can be transferred by the agency of work or of heat, the internal energy remains constant. This law is a cousin of the law of the conservation of energy in mechanics, but it is richer, for it implies the equivalence of heat and work for bringing about changes in the internal energy of a system (and heat is foreign to classical mechanics). The first law is a statement of permission: no change may occur in an isolated system unless that change corresponds to a constant internal energy. See also: Conservation of energy
It is common in thermodynamics to switch attention from the change in internal energy of a system to the change in enthalpy, ΔH, of the system. The change in internal energy and the change in enthalpy of a system subjected to constant pressure p are related by Eq. (2),
where ΔV is the change in volume of the system that accompanies the change of interest. The interpretation of ΔH is that it is equal to the energy that may be obtained as heat when the process occurs. This interpretation follows from the fact that the term pΔV takes into account the work of driving back the surroundings that must take place during the process, and that is therefore not available for providing heat. Enthalpy changes are widely used in thermochemistry, the branch of physical chemistry concerned with the heat productions and requirements of chemical reactions. See also: Enthalpy; Thermochemistry
The enthalpy itself is defined in terms of the internal energy by Eq. (3).
Two important quantities in thermodynamics are the heat capacities at constant volume Cv and constant pressure Cp. These quantities are defined as the slope of the variation of internal energy and enthalpy, respectively, with respect to temperature. The two quantities differ on account of the work that must be done to change the volume of the system when the constraint is that of constant pressure, and then less energy is available for raising the temperature of the system. For a perfect gas, they are related by Eq. (4),
where n is the amount of substance and R is the gas constant. See also: Gas; Heat capacity; Pressure
Second law of thermodynamics
The second law of thermodynamics deals with the distinction between spontaneous and nonspontaneous processes. A process is spontaneous if it occurs without needing to be driven. In other words, spontaneous changes are natural changes, like the cooling of hot metal and the free expansion of a gas. Many conceivable changes occur with the conservation of energy globally, and hence are not in conflict with the first law; but many of those changes turn out to be nonspontaneous, and hence occur only if they are driven.
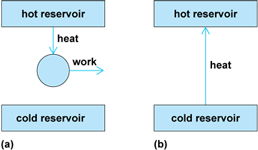
The second law was formulated by Irish physicist William Thomson (Lord Kelvin) and by German theoretical physicist Rudolf Clausius in a manner relating to observation: “no cyclic engine operates without a heat sink” and “heat does not transfer spontaneously from a cool to a hotter body,” respectively (Fig. 3). The two statements are logically equivalent in the sense that failure of one implies failure of the other. However, both may be absorbed into a single statement: the entropy of an isolated system increases when a spontaneous change occurs. The property of entropy is introduced to formulate the law quantitatively in exactly the same way that the properties of temperature and internal energy are introduced to render the zeroth and first laws quantitative and precise.
The entropy, S, of a system is a measure of the quality of the energy that it stores. The formal definition is based on Eq. (5),
where dS is the change in entropy of a system, dq is the energy transferred to the system as heat, T is the temperature, and the subscript “reversible” signifies that the transfer must be carried out reversibly (without entropy production other than in the system). When a given quantity of energy is transferred as heat, the change in entropy is large if the transfer occurs at a low temperature and small if the temperature is high.
This somewhat austere definition of entropy is greatly illuminated by Austrian theoretical physicist Ludwig Boltzmann's interpretation of entropy as a measure of the disorder of a system. The connection can be appreciated qualitatively at least by noting that if the temperature is high, the transfer of a given quantity of energy as heat stimulates a relatively small additional disorder in the thermal motion of the molecules of a system; in contrast, if the temperature is low, the same transfer could stimulate a relatively large additional disorder. This connection between entropy and disorder is justified by a more detailed analysis, and in general it is safe to interpret the entropy of a system or its surroundings as a measure of the disorder present.
The formal statement of the second law of thermodynamics is that the entropy of an isolated system increases in the course of a spontaneous change. The illumination of the law brought about by the association of entropy and disorder is that in an isolated system (into which technology cannot penetrate) the only changes that may occur are those in which there is no increase in order. Thus, energy and matter tend to disperse in disorder (that is, entropy tends to increase), and this dispersal is the driving force of spontaneous change. See also: Arrow of time
This collapse into chaos need not be uniform. The isolated system need not be homogeneous, and there may be an increase in order in one part so long as there is a compensating increase in disorder in another part. Thus, in thermodynamics, collapse into disorder in one region of the universe can result in the emergence of order in another region. The criterion for the emergence of order is that the decrease in entropy associated with it is canceled by a greater increase in entropy elsewhere. See also: Entropy
Third law of thermodynamics
The practical significance of the second law is that it limits the extent to which the internal energy may be extracted from a system as work. In short, in order for a process to generate work, it must be spontaneous. (There is no point in using a process to produce work if that process itself needs to be driven; it is then no more than a gear wheel.) Therefore, any work-producing process must be accompanied by an increase in entropy. If a quantity of energy were to be withdrawn from a hot source and converted entirely into work, there would be a decrease in the entropy of the hot source, and no compensating increase elsewhere. Therefore, such a process is not spontaneous and cannot be used to generate work. For the process to be spontaneous, it is necessary to discard some energy as heat in a sink of lower temperature. In other words, nature in effect exacts a tax on the extraction of energy as work. There is therefore a fundamental limit on the efficiency of engines that convert heat into work (Fig. 4).
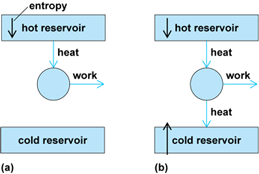
The quantitative limit on the efficiency, ∊, which is defined as the work produced divided by the heat absorbed from the hot source, was first derived by French physicist Sadi Carnot. He found that, regardless of the details of the construction of the engine, the maximum efficiency (that is, the work obtained after payment of the minimum allowable tax to ensure spontaneity) is given by Eq. (6),
where Thot is the temperature of the hot source and Tcold is the temperature of the cold sink. The greatest efficiencies are obtained with the coldest sinks and the hottest sources, and these are the design requirements of modern power plants. See also: Carnot cycle
Perfect efficiency (∊ = 1) would be obtained if the cold sink were at absolute zero (Tcold = 0). However, the third law of thermodynamics, which is another summary of observations, asserts that absolute zero is unattainable in a finite number of steps for any process. Therefore, heat can never be completely converted into work in a heat engine. The implication of the third law in this form is that the entropy change accompanying any process approaches zero as the temperature approaches zero. That implication in turn implies that all substances tend toward the same entropy as the temperature is reduced to zero. It is therefore sensible to take the entropy of all perfect crystalline substances (substances in which there is no residual disorder arising from the location of atoms) as equal to zero. A common short statement of the third law is therefore that all perfect crystalline substances have zero entropy at absolute zero (T = 0). This statement is consistent with the interpretation of entropy as a measure of disorder, since at absolute zero all thermal motion has been quenched. See also: Absolute zero
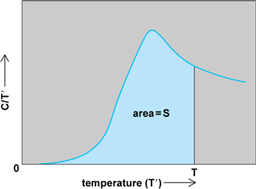
In practice, the entropy of a sample of a substance is measured by determining its heat capacity, C, at all temperatures between zero and the temperature of interest, T, and evaluating the integral given in Eq. (7).
Graphically, C/T′ is plotted against T′, and the area under the curve up to the temperature of interest is equal to the entropy (Fig. 5). In practice, measurements of the heat capacity are made down to as low a temperature as possible, and certain approximations are generally carried out in order to extrapolate these measurements down to absolute zero. A polynomial is fitted to the data, and the integration in Eq. (7) is performed analytically. If there are phase transitions below the temperature of interest, a contribution from each such transition is added, equal to the enthalpy change of the transition divided by the temperature at which it occurs. Such determinations show that the entropy of a substance increases as it changes from a solid to a liquid to a gas. See also: Phase transitions
Gibbs free energy
One of the most important derived quantities in thermodynamics is the Gibbs energy, G, which is widely called the free energy and was formulated by U.S. physicist Willard Gibbs. It is defined by Eq. (8),
where H is the enthalpy of the system, T is its thermodynamic temperature, and S is its entropy. The principal significance of G is that a change in G is a measure of the energy of the system that is free to do work other than simply driving back the walls of the system as the process occurs. For instance, it is a measure of the electrical work that may be extracted when a chemical reaction takes place, or the work of constructing a protein that a biochemical process may achieve. See also: Protein
The Gibbs energy can be developed in two different ways. First, it is quite easy to show from formal thermodynamics that Eq. (9)
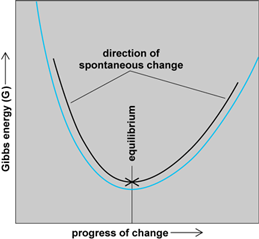
is valid. That is, the change, ΔG, in the Gibbs energy is proportional to the total change, ΔS(total), in the entropy of the system and its surroundings. The negative sign in Eq. (9) indicates that an increase in the total entropy corresponds to a decrease in the Gibbs energy (Fig. 6). Because a spontaneous change occurs in the direction of the increase in total entropy, it follows that another way of expressing the signpost of spontaneous change is that it occurs in the direction of decreasing Gibbs energy. To this extent, the Gibbs energy is no more than a disguised version of the total entropy.
However, the Gibbs energy is much more than that, for (as discussed above) it shows how much nonexpansion work may be extracted from a process. Broadly speaking, because a change in Gibbs energy at constant temperature can be expressed as ΔG = ΔH – TΔS, the latter term represents the tax exacted by nature to ensure that overall a process is spontaneous. Whereas ΔH measures the energy that may be extracted as heat, some energy may need to be discarded into the surroundings to ensure that overall there is an increase in entropy when the process occurs, and that quantity (TΔS) is then no longer available for doing work. This is the origin of the name free energy for the Gibbs energy, for it represents that energy stored by the system that is free to be extracted as work.
For a chemical reaction, the standard reaction Gibbs energy is calculated from the differences of the standard Gibbs energies of formation of each substance, the change in Gibbs energy accompanying the formation of the substance from its elements under standard conditions (a pressure of 1 bar or 105 pascals). The standard reaction Gibbs energy is the principal thermodynamic function for applications in chemistry.
The Gibbs energy is at the center of attention in chemical thermodynamics. It is deployed by introducing a related quantity called the chemical potential. The chemical potential is defined as the slope of the graph showing how the total Gibbs energy of a system varies as the composition of one of its components is increased. The slope of the graph varies with composition, so the chemical potential also varies with composition. Broadly speaking, the chemical potential can be interpreted as a measure of the potential of a substance to undergo chemical change: if its chemical potential is high, then it has a high potential for bringing about chemical change. Thus, a gas at high pressure has a higher chemical potential than one at low pressure, and a substance at high temperature has a higher chemical potential than the same substance at a lower temperature.
An implication of the second law is that the chemical potential of a substance must be the same throughout any phase in which it occurs and the same in all phases that are at equilibrium in a system. These requirements lead, by a subtle argument, to one of the most celebrated conclusions in chemical thermodynamics, the Gibbs phase rule, Eq. (10).
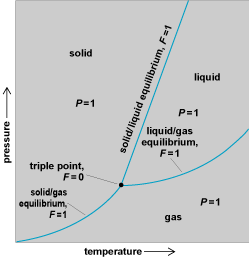
In this expression, C is the number of components in the system (essentially the number of chemically distinct species), P is the number of phases that are in equilibrium with one another, and F is the variance, the number of variables that may be changed without changing the number of phases that are in equilibrium with one another. (In some formulations, C is denoted S − M, where S is the number of substances and M is the number of reactions which relate them and which are at equilibrium.) The phase rule is particularly important for discussing the structure of phase diagrams, which are charts showing the range of temperature and composition over which various phases of a system are in equilibrium (Fig. 7). See also: Phase equilibrium; Phase rule
In systems in which chemical reactions can occur, the chemical potentials of the reactants and products can be used to determine the composition at which the reaction mixture has reached a state of dynamic equilibrium, with no remaining tendency for spontaneous change. In chemistry, the state of chemical equilibrium is normally expressed in terms of the equilibrium constant, K, which is defined in terms of the concentrations or partial pressures of the participating species. In general, if the products dominate in the reaction mixture at equilibrium, then the equilibrium constant is greater than 1, and if the reactants dominate, then it is less than 1. Manipulations of standard thermodynamic relations show that the standard reaction Gibbs energy of any reaction is proportional to the negative of the logarithm of the equilibrium constant. See also: Chemical equilibrium; Chemical thermodynamics; Free energy
Thermodynamics of irreversible processes
The thermodynamic study of irreversible processes centers on the rate of entropy production and the relation between the fluxes and the forces that give rise to them. These fluxes include diffusion (the flux of matter), thermal conduction (the flux of energy of thermal motion), and electrical conduction (the flux of electric charge). In each case, the flux arises from a generalized potential difference of some kind. Thus, diffusion is related to a concentration gradient, and thermal conduction is related to a temperature gradient. In each case, the rate of change of entropy arising from the flux is proportional to both the flux and the gradient giving rise to the flux. Thus, a high flux of matter down a steep concentration gradient results in a rapid change in entropy. See also: Conduction (electricity); Diffusion; Heat conduction
An important observation is that the fluxes, Ji, and potentials, Xj, are not independent of one another. Thus, a temperature gradient can result in diffusion, and a concentration gradient can result in a flux of energy. The general relation between flux and potential is therefore given by Eq. (11),
where the Lij are called the phenomenological coefficients. It was shown by Norwegian-born U.S. theoretical chemist Lars Onsager that for conditions not far from equilibrium, Lij = Lji. This Onsager reciprocity relation implies that there is a symmetry in the ability of a potential Xj to give to a flux Ji and of a potential Xj to give rise to a flux Jj. See also: Thermodynamic processes; Thermoelectricity

